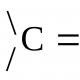Молекулярні діапазони. Коливальні спектри двоатомних молекул Хімія коливальні спектри елемента
Типи коливань
Енергія, необхідна для порушення коливань атомів у молекулі, відповідає енергії квантів світла з довжиною хвилі 1-15 мкм або хвильовим числом 400÷4000 см -1 , тобто електромагнітного випромінювання середньої інфрачервоної області. Коливальні рівні молекул квантовані, енергія переходів між ними і, отже, частоти коливань можуть мати лише певні значення. Поглинаючи квант світла, молекула може переходити більш високий коливальний рівень, зазвичай з основного коливального стану збуджений. Поглинена енергія потім передається на збудження обертальних рівнів або перетворюється на кінетичну енергію молекул. Коливання молекул виявляються у двох типах спектрів: спектри поглинання в інфрачервоній області (ІЧ-спектри) та спектри комбінаційного розсіювання світла (спектри КР).
Математична модель коливань багатоатомних молекул є складною. Розрахунки проведені лише для найпростіших двоатомних молекул. Коливальна спектроскопія має переважно емпіричний характер, тобто. основні частоти коливань отримані зі зіставленням спектрів багатьох сполук одного класу. Це, однак, не применшує цінності методу.
Основними типами коливань є валентні та деформаційні.
Валентнимиколиваннями називаються коливання ядер атомів вздовж лінії зв'язку, вони позначаються буквою n (n C = C , n C = O і т.д.).
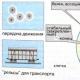
Наближеною механічною моделлю валентних коливань може бути система із двох куль, пов'язаних жорсткою пружиною (тут кулі зображають атоми, а пружина - хімічний зв'язок) (див. рис., а).
А, В – валентні коливання у молекулах;
С - деформаційні коливання: I, II - ножичні; III,IV – маятникові; V – віялові; VI – крутильні.
При розтягуванні або стиску пружини кулі почнуть коливатися навколо положення рівноваги, тобто буде здійснюватися гармонійне коливання, що описується рівнянням

де n - Частота коливання; F - силова постійна, що характеризує міцність зв'язку, або силу, що повертає кулі у положення рівноваги; m r - наведена маса куль (атомів), що обчислюється за формулами

Частоти валентних коливань визначаються масою атомів та міцністю (енергією) зв'язку. Чим маса більша, тим менша частота, наприклад:
n C-C» 1000 см-1; n C-Н» 3000 см -1
Чим зв'язок міцніший, тим вище частота коливань, наприклад:
Можлива поява обертонів - коливань, частота яких більша в ціле число разів, ніж у основних ( 2n, 3n і т.д.). Зазвичай інтенсивність обертонів набагато менше: для першого обертону вона становить 1-10 % від інтенсивності основного коливання; Третій обертон виявити зазвичай не вдається.
У системі з трьох або чотирьох атомів можливі два типи валентних коливань - синфазний (в одній фазі, або симетричний, n s ) і антифазне (у різних фазах, або антисиметричне, n as ) (рис. У), хоча терміни цілком застосовні й у симетричних молекул. Частота антифазного коливання завжди вище, ніж синфазного.
Деформаційніколивання пов'язані із зміною валентного кута, утвореного зв'язками у загального атома; вони позначаються буквою d . Види деяких деформаційних коливань показані на рис., З. Для порушення деформаційних коливань потрібна менша енергія, ніж у разі валентних коливань, і, отже, вони мають меншу частоту.
Зі збільшенням числа атомів у молекулі число можливих коливань швидко зростає. У реальній молекулі коливання атомів тісно пов'язані між собою і взаємодіють між собою. Спектри молекул є складним набором різних коливань, кожне з яких проявляється у вузькому інтервалі частот.
Інтенсивність поглинання визначається, як і в УФ спектроскопії, молярним коефіцієнтом поглинання, однак у цьому випадку точність виміру істотно менша. Зазвичай інтенсивність смуг виражають як поглинання (А) або пропускання (Т) світлового потоку в процентах. Смуги також оцінюють за інтенсивністю як сильні ( с.), середні ( пор.) та слабкі ( сл.).
Отримання ІЧ спектрів
В основі отримання ІЧ спектрів лежить пряме поглинання випромінювання під час проходження через шар речовини. З великого діапазону ІЧ випромінювання зазвичай використовують середню область (400-4000 см -1). В області ближнього ІЧ (4000÷14300 см -1), де виявляються переважно обертони, проводять іноді кількісний аналіз. У далеку ІЧ-область (100÷400 см -1) потрапляють майже лише коливання зв'язків вуглець-метал.
Схема ІЧ спектрометра подібна до схеми УФ спектрометра, але система приладів більш складна. ІЧ випромінювання є тепловим; його джерелом зазвичай служить керамічний стрижень, що розжарюється проходить електричним струмом. За допомогою системи дзеркал світловий потік поділяється на два однакові промені, один з яких пропускається через кювету з речовиною, інший - через кювету порівняння. Пройшло через кювети випромінювання надходить в монохроматор, що складається з призми, дзеркала і щілини, що обертається, що дозволяє виділяти випромінювання зі строго визначеною частотою і плавно змінювати цю частоту. Враховуючи, що в ІЧ області більшість речовин непрозора, призми виготовляють із монокристалів солей. У приладах високого класу застосовують три призми: LiF(2000÷3800 см -1), NaCl(700÷2000 см -1) та КВr(400÷700 см -1). Кожна із призм в іншому інтервалі хвильових чисел дає значно менший дозвіл. У ряді приладів дисперсія випромінювання здійснюється за допомогою дифракційних ґрат. Інтенсивності двох світлових потоків (основного та променя порівняння), що пройшли через монохроматор, автоматично віднімаються одна з іншої. Електричний сигнал, що утворюється при попаданні результуючого світлового потоку на детектор типу термопари, посилюється та реєструється самописним потенціометром. Запис є ІЧ спектр у вигляді залежності поглинання або пропускання (в %) від частоти (см -1) або довжини хвилі (мкм). Типовий вид спектра представлено на рис.

ІЧ спектри найчастіше отримують таким чином:
1. Розчини речовиннайбільш зручні для отримання спектрів, тому що в цьому випадку відсутні міжмолекулярні взаємодії. У зв'язку з тим, що в ІЧ області поглинає будь-яку речовину, як розчинники використовують сполуки найпростішої структури, спектр яких має найпростіший вид (мінімальна кількість смуг), і найчастіше - чотирихлористий вуглець, який прозорий вище 1300 см -1 , а також сірковуглець , практично прозорий та нижче 1300 см -1 . Послідовно розчинивши речовину в тому й іншому розчиннику, вдається записати весь інфрачервоний спектр.
Для розчинів застосовують циліндричні кювети товщиною 0,1 ÷ 1,0 мм із вікнами з сольових пластин. Необхідний для заповнення кювети об'єм розчину 0,1 - 1,0 мл при концентрації 0,05 - 10%.
2. Тонкі плівки (<0,01 мм) жидкого вещества, помещенные между солевыми пластинами, удерживаемыми капиллярными силами.
3. Пасти, що готуються ретельним розтиранням твердого зразка з вазеліновим маслом і поміщаються у вигляді тонкого шару між сольовими пластинами. Саме вазелінове масло, що є сумішшю вуглеводнів, інтенсивно поглинає в області 2900 см -1 і 1400 см -1 . Іноді для приготування паст використовується гексахлорбутадієн, прозорий вище 1600 см -1 і області 1250÷1500 см -1 , тобто в тих інтервалах частот, в яких поглинає вазелінове масло.
4. Тверді речовини у вигляді тонкого порошку(0,5÷1,0 мг), ретельно перемішані з порошком броміду калію (~100 мг) і потім спресовані у спеціальному пристрої під тискомдо »4,5×10 8 Па тонку пластину.
5. Метод порушеного повного внутрішнього відображення(НПО):

Кількість речовини, необхідне отримання ІК спектру, незалежно від способу приготування проби становить 0,5÷2 мг.
Оскільки матеріалом для кювету є сольові пластини, зразок не повинен містити води. Метод ІЧ-спектроскопії - один із найбільш доступних у лабораторній практиці. Прилади прості в обігу, для отримання спектра потрібно кілька хвилин.
Іншим типом спектрів, що несе інформацію про коливання в цьому діапазоні, є спектри комбінаційного розсіювання (КР).

Основною особливістю їх є фіксація довжин хвиль переважно у видимому діапазоні. Умовою їх отримання є наявність високоінтенсивного джерела високомонохроматичного випромінювання, частіше лазера, а спочатку окремих ліній атомного спектру люмінесцентної ртутної лампи низького тиску.
Спектр утворюється внаслідок непружних взаємодій фотонів світлового пучка з молекулами речовини. Фотон, стикаючись з електроном молекули, може переводити його більш високий молекулярний енергетичний рівень, втрачаючи у своїй частина енергії. Лінії, що з'являються стоксовими . Можливий випадок, коли фотон стикається з електроном, що знаходиться на високому молекулярному енергетичному рівні, і переводить його на нижчу орбіталь, захоплюючи частину енергії. З'являються лінії, симетричні стоксовим відносно основної лінії (фотон, що падає) і звані антистоксовими . Стоксівські лінії, тобто. менш енергійні, інтенсивніші, т.к. процес передачі енергії фотоном електрону вірогідніший. Однак всі лінії КР-спектрів малоінтенсивні в порівнянні з збуджуючої (всього близько 10 -7 від загальної інтенсивності розсіяного світла). Тому КР спектри фіксують перпендикулярно напрямку збудливого пучка. Реєстрацію спектра здійснюють як завжди. При цьому біля основної збудливої лінії n 0 утворюється ряд вузьких ліній, що відповідають n i . По відстанях між n 0 і n i визначаються величини Dn .
Вид спектру подібний до одержуваного в ІЧ спектроскопії. У сучасних приладах збудження розсіяного світла виробляється монохроматичним лазерним променем, що дозволяє при отриманні діапазону обходитися 1÷10 мг речовини. Пробу можна вводити як чистої рідини або розчину, так і у вигляді твердого порошку.
Електромагнітні коливання виникають з допомогою руху зарядів. Відповідно, їхнє поглинання пов'язане зі зміщенням зарядів. Очевидно, що пряме поглинання в ІЧ області відбуватиметься з достатньою інтенсивністю, якщо зв'язок полярний. У спектрах КР інтенсивні смуги дають симетричні коливання неполярних зв'язків, оскільки у разі важливий дипольний момент, що у процесі коливання. Отже, у найпростіших випадках в ІЧ спектрах повинні проявлятися коливання, неактивні у спектрах КР, і, відповідно, навпаки. Для симетричних молекул в ІЧ спектрах активні антифазні коливання, тоді як у спектрах КР – синфазні. У міру зниження симетрії молекули багато коливань досить інтенсивно виявляються в тому та іншому спектрі. Отже, ІЧ і КР спектри доповнюють один одного, і при спільному застосуванні цих методів може бути отримана максимальна інформація про частоти коливань досліджуваної речовини.
Смуги в коливальних діапазонах поділяються на два типи. Характеристичні(переважно валентні) смуги, присутність яких у спектрі доводить наявність у досліджуваній речовині певних структурних елементів.
Характеристичними є ті коливання, які хоча б за одним параметром (m r або F ) істотно відрізняються від основних коливань С-С (це коливання легких атомів: С-Н, О-Н, N-Н чи кратних зв'язків).
Характеристичне коливання належить до певного зв'язкуі, отже, має досить постійну частоту в різних речовинах, яка змінюється лише трохи за рахунок взаємодії з рештою молекули.
Нехарактеристичнісмуги, що займають область 400÷1000 см -1 , де виявляються численні валентні коливання зв'язків, що не піддаються віднесенню С-С, С-N, N-О та деформаційні коливання. Це область коливань вуглецевого скелета молекули, яка різко реагує на найменші зміни у структурі молекули. Нехарактеристичні коливання становлять основну частину спектра і кожної речовини утворюють свій, неповторний набір смуг. Немає двох сполук за винятком енантіомерів (оптичних антиподів), які мали б однакові інфрачервоні спектри (і спектри КР). Цим часто користуються для встановлення тотожності речовин, оскільки збіг ІЧ спектрів є переконливим доказом ідентичності досліджуваних зразків.
Враховуючи, що в спектрі однієї речовини завжди може бути знайдена смуга, яка відсутня в спектрі іншої, можливий якісний аналіз сумішей, якщо спектри компонентів відомі.
На тій самій підставі може бути виконаний кількісний аналіз шляхом вимірювання інтенсивностей відповідних смуг. Коли будова речовини вже встановлено, у нехарактеристичній області спектра деякі смуги вдається віднести до певних коливань.
Однак перед дослідником стоїть зазвичай протилежне завдання – встановити будову за спектром. В цьому відношенні можливості ІЧ спектроскопії не потрібно переоцінювати, слід використовувати лише абсолютно надійні критерії.
Зокрема, дані, отримані з розгляду області нижче 1500 см -1 , не можна розцінювати як докази, лише як свідчення на користь присутності того чи іншого структурного елемента. До помилкових висновків може призвести використання структурних віднесень (зокрема, визначення конформації і найближчого оточення) малих змін у величині характеристичної частоти.
Іншими словами, з коливальних спектрів не слід намагатися отримати інформацію, достовірність якої є сумнівною.
Для опису коливальних спектрів найчастіше використовують таку інформацію:
Коливання зв'язку С-Н. Валентні коливання С-Н при насиченому вуглецевому атомі виявляються в області 2800-3000 см -1 . Для ациклічних та ненапружених циклічних структур n CH мають наступні значення (см -1):
| СН 3 | 2962 см -1 | 2972 см -1 |
| СН 2 | 2853 см -1 | 2926 см -1 |
| СН | 2890 см -1 |
Смуги характерні, але малоінформативні, тому що в речовині зазвичай спостерігаються різні коливання С-Н , які, крім того, можуть взаємодіяти між собою. Окремі смуги коливань накладаються одна на одну, утворюючи в області 2800÷3000 см -1 смугу, що має окремі слабко виражені максимуми. Для визначення структури речовини ці смуги можуть бути корисними тільки в тому випадку, якщо в поєднанні мало атомів водню, як, наприклад, полігалогеналканах. Відсутність смуг у цій галузі є переконливим доказом відсутності у речовині атомів водню при насичених вуглецевих атомах.
Деформаційні коливання d СН , розташовані в області 1350÷1470 см -1 , малохарактеристичні, але можуть бути виявлені в спектрі:
| СН 3 | 1375 см -1 | 1450 см -1 |
| СН 2 | 1475 см -1 |
Досить характерним вважається поглинання двох метильних груп при одному вуглецевому атомі (гемінальне заміщення), що утворює два близькі максимуми (дублет) приблизно рівної інтенсивності в області 1370÷1385 см -1 .
Інформативність спектрів можна підвищити, використовуючи відмінності в частотах коливань зв'язків, що містять різні ізотопні модифікації атомів. Зокрема, часто використовують дейтеровані сполуки, збагачені дейтерієм замість протию.
При аналізі сполук, мічених дейтерієм, дуже характерною є смуга n CD 2100÷2160 см -1 розташована в області, де практично відсутні інші смуги.
Коливання зв'язку С=С. У з'єднаннях із ізольованим подвійним зв'язком v c = c знаходиться при 1600-1680 см -1 .
У циклічних системах, особливо напружених, значення цієї частоти трохи нижче. Частота коливань подвійного зв'язку помітно підвищується зі зростанням ступеня її заміщеності, наприклад:

В ІЧ спектрах симетрично заміщених алкенів (неполярний подвійний зв'язок) n С=С проявляється смугою мізерно малої інтенсивності, як, наприклад, у спектрах сполук (I) та (III); для несиметрично заміщеного подвійного зв'язку (наприклад, з'єднання II) ця смуга досить інтенсивна. У спектрах КР коливання С=С у разі активніше, ніж у ІЧ спектрі, і будь-яка подвійна зв'язок дає потужну (зазвичай найбільш інтенсивну у спектрі) лінію. Про наявність речовини подвійного зв'язку додатково може свідчити характеристична смуга (смуги) n = CH , розташована в області 3000-3100 см -1 .
Деформаційні коливання d =СН можуть бути корисні для визначення конфігурації заступників при подвійному зв'язку: для цис-ізомерів вони розташовані в області 650÷750 см -1 , а для транс-ізомерів - в області 960÷970 см -1 .
Таким чином, на підставі даних коливальних спектрів (особливо спектра КР) може бути виявлено присутність у речовині ізольованого подвійного зв'язку та зроблено певні висновки про характер її заміщення.
Смуга n =С D дуже характеристична (2200÷2300 см -1) і дозволяє впевнено відрізнити атом дейтерію, що знаходиться при подвійному зв'язку, від атома D при насиченому вуглецевому атомі.
Коливання пов'язаних дієнових систем.
Сполучені дієнові системи в області 1500÷1650 см -1 мають дві смуги, що відповідають двом типам валентних коливань - синфазному та антифазному, наприклад: 
У цілому нині смуги коливань дієнових систем в ІЧ- і КР- спектрах значно інтенсивніші проти смугами ізольованих подвійних зв'язків, якщо дієнова система має трансоїдну конфігурацію. В ІЧ спектрі більш активно коливання, тоді як у спектрі КР-коливання. В інфрачервоному спектрі симетричних дієнів (наприклад, бутадієну) інтенсивність смуги може бути зникаюче мала. При введенні в дієнову систему алкільних замісників значення частот і підвищуються закономірно. Коливання n = CH у дієнах виявляються у тій самій області, що й у алкенах (3000÷3100 див -1).
Таким чином, наявність у речовині дієнової системи відносно легко визначається за даними коливальних спектрів. При поєднанні подвійного зв'язку з ароматичним ядром частота її коливання зміщується в низькочастотну область (на 30 см -1), при цьому інтенсивність поглинання підвищується. При збільшенні довжини ланцюга сполучення (у спектрах полієнів) зростає загальна кількість смуг n С=С , причому частоти їх коливань зменшуються, а інтенсивність значно зростає.
Коливання ароматичних систем. Валентні коливання С-З-зв'язків бензольного ядра дають смуги помірної інтенсивності при 1585-1600 см -1 і 1400-1500 см -1 , що робить їх незручними для ідентифікації, так як ця область близька до коливань n С=С. Коливання n CH аренів лежать в області 3020-3100 см -1; зазвичай вони проявляються у вигляді групи смуг середньої інтенсивності, дещо більшої, ніж у поглинаючих у тій же області n = CH алкенів.
У спектрах ароматичних сполук є інтенсивні смуги непоганих деформаційних коливань С-Н в області 650÷900 см -1. Ця область дає деякі можливості для визначення кількості та розташування замісників в ароматичному ядрі, а також взаємного розташування бензольних кілець у поліядерних ароматичних сполуках. Як правило, відсутність сильних смуг в області 650-900 см -1 свідчить про відсутність у речовині ароматичного ядра. Крім того, у цій галузі проявляються коливання зв'язків вуглець-галоген, причому смуги зазвичай мають високу інтенсивність: C-Cl (550÷850 см -1), C-Br (515÷690 см -1), C-I (500÷600 см -1). Коливання зв'язку C-F виявляються в області скелетних коливань зв'язків С-С тому спостерігати їх дуже складно. Використовувати коливання зв'язків вуглець-галоген для визначення галогенів у складі речовини не має сенсу (є безліч методів швидших і точніших), але для спостереження проміжних продуктів та взаємодій у дослідженні механізмів реакцій поява смуг може дати корисну інформацію.
Для встановлення положення заступників в ароматичному ядрі іноді використовують область 1650÷2000 см -1 де виявляються виключно слабкими смугами обертони і тони більш складного походження. Смуги у цій галузі залежно від характеру заміщення мають різний контур. Надійність цієї ознаки невелика, і, крім того, ця область повністю перекривається за наявності речовини карбонильной групи.
Коливальні спектри найважливіших гетероциклічних систем мають багато спільного зі спектрами похідних бензолу: так, для фурану, тіофену, піролу та піридину n CH 3010÷3080 см -1 та n C-С (кільце) 1300÷1600 см -1 , причому положення смуги v С-С суттєво залежить від типу гетероциклу та характеру заміщення. У цій галузі може виникнути від двох до чотирьох смуг. Нижче наводяться основні частоти в спектрах найважливіших гетероциклів (см -1)
Коливання зв'язку СºС. Наявність зв'язку зазвичай встановлюють смугою валентних коливань 2100÷2250 см -1 , т.к. у цій галузі інші смуги практично відсутні. Смуга середньої інтенсивності, при симетричному заміщенні в ІЧ спектрі вона може стати практично непомітною, у КР спектрі смуга активна завжди і її інтенсивність тим більша, чим менш симетричний алкін.
Коливання зв'язку О-Н. У сильно розведених розчинах, що забезпечують відсутність міжмолекулярних взаємодій, гідроксильні групи проявляються високоінтенсивною смугою валентних коливань 3200-3600 см-1. Якщо гидроксогруппа бере участь у водневого зв'язку, то становище та характер лінії починає сильно залежати від ступеня залученості, т.к. починає змінюватися силовий постійний зв'язок. Якщо зв'язок міжмолекулярний, з'являється широка неструктурована смуга, що перекриває весь діапазон 3200÷3600 см -1 . Якщо спостерігається внутрішньомолекулярний водневий зв'язок, про це свідчить інтенсивна смуга близько 3500 см -1 , зміщена в область низьких частот порівняно з вільними групами. Для уникнення можливості утворення міжмолекулярних зв'язків слід використовувати малополярні розчинники (вуглеводні, CCl 4) та концентрацію менше 5×10 -3 моль/л. Вільний фенольний гідроксил проявляється смужкою валентного коливання 3600÷3615 см -1 високої інтенсивності.
Деформаційні коливання гідроксогруп розташовані в області 1330-1420 см -1 і малопридатні для ідентифікації. Димери карбонових кислот виявляються широкою інтенсивною смугою в області 1200÷1400 см -1 але віднесення смуги може бути впевнено зроблено тільки після доказу, що речовина дійсно карбонова кислота.
Коливання зв'язку С-О. Зв'язок проявляється у простих ефірах та спиртах інтенсивною смугою області 1000÷1275 см -1 . Складні ефіри в спектрах містять дві смуги за рахунок групи С-О-С: симетричного коливання 1020÷1075 (слабша в ІЧ спектрі) і антисиметричного при 1200÷1275 см -1 (слабша в КР спектрі). У цьому діапазоні проявляються смуги різних груп та смуги малохарактеристичні, але найчастіше саме вони найінтенсивніші.
Коливання зв'язку З=О. Валентні коливання карбонільної групи присутні у спектрах різних сполук: альдегідів, кетонів, карбонових кислот, ангідридів тощо. Це завжди високоактивний пік області 1650÷1680 см -1 , де інші смуги практично відсутні. Це одна з найбільш характеристичних смуг, її наявність або відсутність може бути переконливим доказом наявності або відсутності карбонільних груп. Конкретний діапазон прояву смуги залежить від сусідніх груп і групи, в яку входить карбоніл, індукційний ефект (-I) зменшує довжину зв'язку С=О і, отже, збільшується силова постійна та частота. Для альдегідів і кетонів смуга знаходиться близько 1710÷1750, карбонових кислот - 1650÷1695,18 - 1865÷1875, ангідридів кислот - 1740÷1790 та 1800÷1850 см -1 . Ефект сполучення p-електронів знижує частоту коливання: в системах С=С-С=О та С 6 Н 5 -С=Про смуга розташована близько 1665÷1685 см -1 .
Таким чином, спектри карбонільних сполук дозволяють отримати великий обсяг цілком однозначної інформації, особливо з огляду на інші смуги: для складних ефірів та ангідридів - смуга С-О , амідів - смуга N-H , у спектрах альдегідів часто присутня смуга групи С(О)-Н близько 2695÷2830 см -1 . Для багатьох альдегідів та кетонів спектр являє собою суму основної та енольної форми.
Зведення спектральних проявів різних угруповань в ІЧ- та КР-спектрах наведено в таблиці №2, проте існують спеціальні таблиці, що містять більший набір частот і дозволяють досліджувати практичні набори смуг від різних зразків.
Таблиця №2 Основні частоти коливань в ІЧ-спектроскопії
| Частота см -1 | Інтенсивність | Природа коливань | Тип з'єднань | |
| 3620- 3600 | с., порівн. | n ВІН (своб.) | Розведені розчини спиртів | |
| 3600- 3500 | с., порівн. | n ВІН (зв.) | Внутрішньомолекулярні водневі зв'язки у спиртах | |
| с., порівн. | (Вільн.) | Розведені розчини первинних амідів | ||
| 3400- 3350 | пор. | n NH (свобод.) | Вторинні аміни, N-заміщені аміди | |
| 3550- 3520 | с., порівн. | n OH (своб.) | Розведені розчини кислот | |
| 3500- 3400 | с., порівн. | n NH2 (свобод.) | Первинні аміни, аміди | |
| с. | (Вільн.) | Розбавлені розчини амідів | ||
| 3330- 3260 | пор. | n º CH | Однозаміщені алкіни | Алкани |
| 1370- 1390 | с., порівн. | Нітросполуки | ||
| 1280- 1200 | с. | n СОС | Складні ефіри | |
| 1250- 1180 | пор. | n С-N | Третичні аміни (ArNR 2 , (RCH 2) 3 N) | |
| 1220- 1125 | с. | n С-О | Вторинні, третинні спирти | |
| 1200- 1160, 1145- 1105 | с., порівн. | n С-О | Кеталі, ацеталі | |
| 1150- 1050 | с. | Ефіри | ||
| 1085- 1050 | с., порівн. | n С-О | Спирти | |
| 970- 950 | пор. | d СН | Транс-алкени | |
| 900-650 | с. | d СН | Арени | |
| 750- 650 | пор. | d =СН | Цисдієни |
| Тип зв'язку та з'єднань | Частота см -1 | ||
| -С=С- | |||
| алкени | 1680- 1620 | ||
| цис-похідні | 1665- 1635 | ||
| транс-похідні | 1675- 1660 | ||
| циклічні | 1650- 1550 | ||
| пов'язані | 1660- 1580 | ||
| -С=С=С- | |||
| алени | 1970-1940 (n as) | ||
| 1070-1060 (n s) | |||
| -CºC- | |||
| Алкіни | 2270- 2190 | ||
| -CºC-H | 2140- 2100 | ||
| ñС=Про | |||
| Кетони | аліфатичні | 1725- 1700 | |
| ненасичені | 1690- 1660 | ||
| арілкетони | 170- 1680 | ||
| діарілкетони | 1670- 1660 | ||
| циклічні | 1780- 1700 | ||
| Дикетони | a | 1730- 1710 | |
| b | 1640- 1635 | ||
| Альдегіди | аліфатичні | 1740- 1720 | |
| ненасичені | 1705- 1650 | ||
| ароматичні | 1715- 1685 | ||
| Карбонові кислоти | мономер | ||
| димер | 1725- 1700 | ||
| ненасичені | 1715- 1680 | ||
| ароматичні | 1700- 1680 | ||
| лактони | 1850- 1720 | ||
| ангідриди | |||
Одночасно зі зміною коливального стану молекули змінюється та його обертальний стан. Зміна коливальних та обертальних станів призводить до виникнення обертально-коливальних спектрів. Коливальна енергія молекул приблизно в сто разів більша за її обертальну енергію, тому обертання не порушує коливальної структури молекулярних спектрів. Накладання невеликих в енергетичному відношенні обертальних квантів на порівняно великі по енергії коливальні кванти, зміщує лінії коливального спектра на ближню інфрачервону область електромагнітного спектра і перетворює їх на смуги. З цієї причини обертально-коливальний спектр, який спостерігається в близькій інфрачервоній ділянці, має лінійно-смугасту структуру.
Кожна смуга такого спектру має центральну лінію (пунктирна лінія), частота якої визначається різницею коливальних термів молекули. Сукупність таких частот є чистим коливальним спектром молекули. Квантово-механічні розрахунки, пов'язані з рішенням хвильового рівняння Шредінгера з урахуванням взаємного впливу обертальних та коливальних станів молекули, призводять до вираження:
де і є постійними всім енергетичних рівнів і залежить від коливального квантового числа.
де - постійні, менші за величиною, ніж і . В силу небагато параметрів і , в порівнянні з величинами і , іншими складовими в цих співвідношеннях можна знехтувати і розглядати власне обертально-коливальну енергію молекули, як суму коливальної та обертальної енергії жорсткої молекули тоді відповідно вираз:
Цей вираз добре передає структуру спектра і призводить до спотворення лише при великих значеннях квантових чисел та . Розглянемо обертальну структуру обертально-коливального спектра. Так, при випромінюванні молекула переходить з вищих енергетичних рівнів і на нижні, і в спектрі з'являються лінії з частотами:
тобто. для частоти лінії обертально-коливального спектра можна записати відповідно:
сукупність частот дає обертально-коливальний спектр. Перший член в даному рівнянні виражає спектральну частоту, що виникає при зміні тільки коливальної енергії. Розглянемо розподіл обертальних ліній у смугах спектра. У межах однієї смуги її тонка обертальна структура визначається лише значенням обертального квантового числа. Для такої смуги можна записати у вигляді:
Відповідно до правила відбору Паулі:
вся смуга поділяється на дві групи спектральних серій, які розташовуються щодо обох боків. Справді, якщо:
тобто. коли:
то отримуємо групу ліній:
тобто. коли:
то отримуємо групу ліній:
У разі переходів коли молекула переходить з - го обертального рівня на обертальний енергетичний рівень, виникає група спектральних ліній із частотами . Ця група ліній називається позитивною або - гілкою смуги спектра, що починається з . При переходах , коли молекула переходить з - го на енергетичний рівень, з'являється група спектральних ліній, з частотами . Ця група ліній називається негативною або - гілкою смуги спектра, що починається з . Це тим, що значення , що відповідає не має фізичного сенсу. - і - гілки смуги, виходячи з рівнянь виду:
складаються з ліній:
Таким чином, кожна смуга обертально-коливального спектру складається з двох груп рівновіддалених ліній з відстанню між сусідніми лініями:
для реальної нежорсткої молекули, враховуючи рівняння:
для частоти ліній - та - гілок смуги, отримуємо:
Внаслідок цього лінії - і - гілок викривляються і спостерігаються не рівновіддалені лінії, а - гілки, що розходяться і - гілки, що зближуються з утворенням канта смуги. Таким чином, квантова теорія молекулярних спектрів виявилася здатною при розшифровці спектральних смуг у ближній інфрачервоній області, трактуючи їх як результат одночасної зміни обертальної та коливальної енергії. Слід зазначити, що молекулярні спектри є цінним джерелом відомостей про будову молекул. Вивчаючи молекулярні спектри, можна безпосередньо визначити різні дискретні енергетичні стани молекул і на підставі отриманих даних зробити надійні та точні висновки щодо руху електронів, коливання та обертання ядер у молекулі, а також отримати точні відомості щодо сил діючих між атомами в молекулах, міжядерних відстанях та геометричній розташування ядер у молекулах, енергії дисоціації самої молекули та ін.
Молекулярні спектри- спектри поглинання, випромінювання або розсіювання, що виникають при квантових переходахмолекул з одного енергетич. стану до іншого. M. с. визначаються складом молекули, її структурою, характером хім. зв'язку та взаємодією з внеш. полями (і, отже, з оточуючими її атомами та молекулами). наиб. характерними виходять M. с. розріджених молекулярних газів, коли відсутня розширення спектральних лінійтиском: такий спектр складається з вузьких ліній з доплерівської шириною.
Рис. 1. Схема рівнів енергії двоатомної молекули: aі б-електронні рівні; u" і u"" - коливальні квантові числа; J"і J"" - обертальні квантові числа.

Відповідно до трьох систем рівнів енергії в молекулі - електронної, коливальної та обертальної (рис. 1), M. с. складаються із сукупності електронних, коливань. і крутять. спектрів і лежать у широкому діапазоні ел-магн. хвиль – від радіочастот до рентг. сфери спектру. Частоти переходів між обертають. рівнями енергії зазвичай потрапляють у мікрохвильову область (у шкалі хвильових чисел 0,03-30 см-1), частоти переходів між колибатами. рівнями -в ІЧ-область (400-10 000 см -1), а частоти переходів між електронними рівнями - у видиму та УФ-області спектра. Цей поділ умовний, тому що часто крутять. переходи потрапляють і в ІЧ-область, коливання. переходи – у видиму область, а електронні переходи – в ІЧ-область. Зазвичай електронні переходи супроводжуються зміною коливань. енергії молекули, а при коливанні. переходах змінюється та обертають. енергія. Тому найчастіше електронний спектр є системою електронно-колебат. смуг, причому при високій роздільній здатності спектральної апаратури виявляється їх обертають. структури. Інтенсивність ліній та смуг у M. с. визначається ймовірністю відповідного квантового переходу. наиб. інтенсивні лінії відповідають переходу, дозволеному відбору правилами. До M. с. відносять також оже-спектри та рентг. спектри молекул(У статті не розглядаються; див. Оже-ефект, Оже-спектроскопія, Рентгенівські спектри, Рентгенівська спектроскопія).
Електронні спектри. Чисто електронні M. с. виникають при зміні електронної енергії молекул, якщо при цьому не змінюються коливання. і крутять. енергії. Електронні M. с. спостерігаються як у поглинанні (спектри поглинання), так і у випромінюванні (спектри). При електронних переходах зазвичай змінюється електрич. . Еле-ктріч. дипольний перехід між електронними станами молекули типу Г "
та Г ""
(Див. Симетрія молекул) дозволено, якщо прямий твір Г "
Г ""
містить тип симетрії, принаймні одного з компонентів вектора дипольного моменту d
. В спектрах поглинання зазвичай спостерігають переходи з основного (повносиметричного) електронного стану в збуджені електронні стани. Очевидно, що для здійснення такого переходу типи симетрії збудженого стану та дипольного моменту мають співпадати. T. к. електрич. дипольний момент не залежить від спина, то при електронному переході спін повинен зберігатися, тобто дозволені лише переходи між станами з однаковою мультиплетністю (інтеркомбінац. заборона). Це правило, однак, порушується
для молекул з сильною спін-орбітальною взаємодією, що призводить до інтеркомбінаційним квантовим переходам. В результаті таких переходів виникають, напр., спектри фосфоресценції, які відповідають переходам з збудженого триплетного стану в осн. синглетний стан.
Молекули у разл. Електронні стани часто мають різну геом. симетрію. У таких випадках умова Г "
Г ""
Г dмає виконуватися для точкової групи низькосиметричної конфігурації. Однак при використанні перестановочно-інверсійної (ПІ) групи така проблема не виникає, тому що ПІ група для всіх станів може бути обрана однаковою.
Для лінійних молекул симетрії З хутип симетрії дипольного моменту Г d= S + (d z)-P( d x , d y), тому їм дозволені лише переходи S + - S + , S - - S - , П - П тощо. буд. з дипольним моментом переходу, спрямованим по осі молекули, і переходи S + - П, П - D тощо. д. з моментом переходу, спрямованим перпендикулярно до осі молекули (позначення станів див. в ст. Молекула).
Ймовірність Велектрич. дипольного переходу з електронного рівня тна електронний рівень п, підсумована по всіх коливально-оберт. рівням електронного рівня т, визначається ф-лою:
матричний елемент дипольного моменту для переходу n - m, y епта y em- хвильові ф-ції електронів. Інтогральний коеф. поглинання, який можна виміряти експериментально, визначається виразом
де N m- Число молекул на поч. стані m, v nm- Частота переходу тп. Часто електронні переходи характеризуються
Автор Хімічна енциклопедія р.н. І.Л.КнунянцКОЛИВАЛЬНІ СПЕКТРИ, мовляв. спектри, зумовлені квантовими переходами між коливальними рівнями енергії молекул. Експериментально спостерігаються як ІЧ спектри поглинання та спектри комбінац. розсіювання (КР); діапазон хвильових чисел ~10-4000 см -1 (частоти коливальних переходів 3 . 10 11 -10 14 Гц). Колебат. рівні енергії визначаються квантуванням коливального руху атомних ядер. Двохтомні молекули. У найпростішому випадку двоатомну молекулу представляють моделлю двох взаємодіючих точкових мас m 1 і m 2 з рівноважною відстанню r е між ними (довжина зв'язку), а коливальний рух ядер вважається гармонічним і описується єдністю, координатою q=rr e , де r - поточна міжядерна відстань . Залежність потенційної енергії коливальний рух V від q визначають у наближенні гармоній. осцилятора [коливання матеріальна точка з наведеною масою m =m 1 m 2 /(m 1 +m 2)] як функцію V= l / 2 (K eq 2), де К е =(d 2 V/dq 2) q= 0 – гармоній. силова постійна 
Рис. 1. Залежність потенційної енергії V гармоній. осцилятора (пунктирна крива) та реальної двоатомної молекули (суцільна крива) від міжядерної відстані r (r з рівноважним значенням r); горизонтальними прямими лініями показані коливальні рівні (0, 1, 2, ... значення коливального квантового числа), вертикальними стрілками - деякі коливальні переходи; D 0 – енергія дисоціації молекули; заштрихована область відповідає суцільному спектру. молекули (пунктирна крива на рис. 1).
Відповідно до класич. механіці, частота гармон. вагань ![]() Квантовомеханічний розгляд такої системи дає дискретну послідовність рівновіддалених рівнів енергії E(v)=hve (v+ 1/2), де v = 0, 1, 2, 3, ... - коливальне квантове число, v e - гармоній. коливальна стала молекули (h - стала Планка). При переході між сусідніми рівнями, згідно з правилом відбору D v=1, поглинається фотон з енергією hv= DE=E(v+1)-E(v)=hv e (v+1+ 1 / 2)-hv e (v+ 1 / 2) = hv e, тобто частота переходу між двома будь-якими сусідніми рівнями завжди одна і та ж, причому збігається з класич. частотою гармоній. коливань. Тому v e називають також гармонічним. частотою. Для реальних молекул крива потенційної енергії не є зазначеною квадратичною функцією q, тобто параболою. Колебат. рівні дедалі більше зближуються з наближенням до межі дисоціації молекули й у моделі ангармонич. Осцилятор описуються рівнянням: E(v)=, де X 1 - перша стала ангармонічності. Частота переходу між сусідніми рівнями не залишається постійною, і, крім того, можливі переходи, що відповідають правилам відбору D v = 2, 3, .... Частота переходу з рівня v = 0 на рівень v = 1 називають основною, або фундаментальною частотою , Переходи з рівня v = 0 на рівні v> 1 дають обертонні частоти, а переходи з рівнів v> 0 - так званої гарячі частоти. В ІЧ спектрі поглинання двоатомних молекул коливальні частоти спостерігаються тільки у гетероядерних молекул (НСl, NO, CO тощо), причому правила відбору визначаються зміною їх електрич. дипольного моменту при коливаннях У спектрах КР коливальні частоти спостерігаються для будь-яких двоатомних молекул, як гомоядерних, так і гетероядерних (N 2 , O 2 , CN і т.п.), так як для таких спектрів правила відбору визначаються зміною поляризуемості молекул при коливаннях. Визначаються з КОЛИВАЛЬНІ СПЕКТРИ с. гармоній. Постійні К е і v e, постійні ангармонічності, а також енергія дисоціації D 0 - важливі характеристики молекули, необхідні, зокрема, для термохімічних розрахунків. Вивчення коливально-обертальних спектрів газів і парів дозволяє визначати обертальне постійні V (див. обертальні спектри), моменти інерції та між'ядерні відстані двоатомних молекул. Багатоатомні молекули розглядають як системи пов'язаних точкових мас. Колебат. рух ядер щодо рівноважних положень при нерухомому центрі мас без обертання молекули як цілого описують зазвичай з використанням так званої внутр. природ. координат q i , що вибираються як зміни довжин зв'язків, валентних та двогранних кутів просторів, моделі молекули. У молекули, що складається з N атомів, є n=3N - 6 (у лінійної молекули 3N - 5) коливальний ступенів свободи. У просторі єств. координат q i складний коливальний рух ядер можна уявити п окремими коливаннями, кожне з певною частотою v k (k приймає значення від 1 до n), з якою змінюються всі природи. координати q i при визначених для цього коливання амплітудах q 0 i і фазах. Такі коливання називають нормальними. Наприклад, триатомна лінійна молекула АХ 2 має три нормальні коливання:
Квантовомеханічний розгляд такої системи дає дискретну послідовність рівновіддалених рівнів енергії E(v)=hve (v+ 1/2), де v = 0, 1, 2, 3, ... - коливальне квантове число, v e - гармоній. коливальна стала молекули (h - стала Планка). При переході між сусідніми рівнями, згідно з правилом відбору D v=1, поглинається фотон з енергією hv= DE=E(v+1)-E(v)=hv e (v+1+ 1 / 2)-hv e (v+ 1 / 2) = hv e, тобто частота переходу між двома будь-якими сусідніми рівнями завжди одна і та ж, причому збігається з класич. частотою гармоній. коливань. Тому v e називають також гармонічним. частотою. Для реальних молекул крива потенційної енергії не є зазначеною квадратичною функцією q, тобто параболою. Колебат. рівні дедалі більше зближуються з наближенням до межі дисоціації молекули й у моделі ангармонич. Осцилятор описуються рівнянням: E(v)=, де X 1 - перша стала ангармонічності. Частота переходу між сусідніми рівнями не залишається постійною, і, крім того, можливі переходи, що відповідають правилам відбору D v = 2, 3, .... Частота переходу з рівня v = 0 на рівень v = 1 називають основною, або фундаментальною частотою , Переходи з рівня v = 0 на рівні v> 1 дають обертонні частоти, а переходи з рівнів v> 0 - так званої гарячі частоти. В ІЧ спектрі поглинання двоатомних молекул коливальні частоти спостерігаються тільки у гетероядерних молекул (НСl, NO, CO тощо), причому правила відбору визначаються зміною їх електрич. дипольного моменту при коливаннях У спектрах КР коливальні частоти спостерігаються для будь-яких двоатомних молекул, як гомоядерних, так і гетероядерних (N 2 , O 2 , CN і т.п.), так як для таких спектрів правила відбору визначаються зміною поляризуемості молекул при коливаннях. Визначаються з КОЛИВАЛЬНІ СПЕКТРИ с. гармоній. Постійні К е і v e, постійні ангармонічності, а також енергія дисоціації D 0 - важливі характеристики молекули, необхідні, зокрема, для термохімічних розрахунків. Вивчення коливально-обертальних спектрів газів і парів дозволяє визначати обертальне постійні V (див. обертальні спектри), моменти інерції та між'ядерні відстані двоатомних молекул. Багатоатомні молекули розглядають як системи пов'язаних точкових мас. Колебат. рух ядер щодо рівноважних положень при нерухомому центрі мас без обертання молекули як цілого описують зазвичай з використанням так званої внутр. природ. координат q i , що вибираються як зміни довжин зв'язків, валентних та двогранних кутів просторів, моделі молекули. У молекули, що складається з N атомів, є n=3N - 6 (у лінійної молекули 3N - 5) коливальний ступенів свободи. У просторі єств. координат q i складний коливальний рух ядер можна уявити п окремими коливаннями, кожне з певною частотою v k (k приймає значення від 1 до n), з якою змінюються всі природи. координати q i при визначених для цього коливання амплітудах q 0 i і фазах. Такі коливання називають нормальними. Наприклад, триатомна лінійна молекула АХ 2 має три нормальні коливання: 
Коливання v 1 називають симетричним валентним коливанням (розтягування зв'язків), v 2 - деформаційним коливанням (зміна валентного кута), v 3 антисиметричним валентним коливанням. У складніших молекулах зустрічаються та інших. нормальні коливання (зміни двогранних кутів, крутильні коливання, пульсації циклів тощо.). Квантування коливальної енергії багатоатомної молекули в наближенні багатовимірного гармонію. осцилятора призводить до сліду, системі коливальних рівнів енергії:
де v ek – гармоній. коливальне постійні, v k - коливальне квантові числа, d k - ступінь виродження рівня енергії по k-му коливальне квантового числа. Осн. частоти в КОЛЮВАЛЬНІ СПЕКТРИ с. обумовлені переходами з нульового рівня [всі v k =0, коливальна енергія на рівні, що характеризуються ![]()
такими наборами квантових чисел v k , в яких тільки одне з них одно 1, а решта рівні 0. Як і у випадку двоатомних молекул, в ангармонич. Наближенні можливі також обертонні та "гарячі" переходи і, крім того, так званої комбіновані, або складові, переходи за участю рівнів, для яких відмінні від нуля два або більше квантових чисел v k (рис. 2). 
Рис. 2. Система коливальних термів E/hc (см»; с - швидкість світла) молекули Н 2 Про і деякі переходи; v 1 , v 2 . v 3 - коливальне квантові числа.
Інтерпретація та застосування. КОЛИВАЛЬНІ СПЕКТРИ с. багатоатомних молекул відрізняються високою специфічністю і представляють складну картину, хоча загальна кількість експериментально спостережуваних смуг може бути істотно меншою від можливого їх числа, що теоретично відповідає передбачуваному набору рівнів. Зазвичай основні частоти відповідають більш інтенсивні смуги в КОЛЕБАТЕЛЬНІ СПЕКТРИ с. Правила відбору та ймовірність переходів в ІЧ та КР спектрах різні, оскільки пов'язані відповідно до змін електрич. дипольного моменту та поляризуемості молекули при кожному нормальному коливанні. Тому поява та інтенсивність смуг в ІЧ та КР спектрах по-різному залежить від типу симетрії коливань (стосунки змін молекули, що виникають у результаті коливань ядер, до операцій симетрії, що характеризують її рівноважну конфігурацію). Деякі зі смуг КОЛИНЕЛЬНІ СПЕКТРИ с. можуть спостерігатися тільки в ІЧ або тільки в КР спектрі, інші – з різною інтенсивністю в обох спектрах, а деякі взагалі експериментально не спостерігаються. Так, для молекул, що не мають симетрії або мають низьку симетрію без центру інверсії, всі основні частоти спостерігаються з різною інтенсивністю в обох спектрах, у молекул з центром інверсії жодна з частот, що спостерігаються, не повторюється в ІЧ і КР спектрах (правило альтернативної заборони); деякі з частот можуть бути відсутніми в обох спектрах. Тому найважливіше із застосувань КОЛИВАЛЬНІ СПЕКТРИ с. - Визначення симетрії молекули зі зіставлення ІЧ і КР спектрів, поряд з використанням ін. Експерим. даних. Задаючись моделями молекули з різною симетрією, можна заздалегідь теоретично розрахувати кожної з моделей, скільки частот в ІЧ і КР спектрах має спостерігатися, і підставі зіставлення з эксперим. даними зробити відповідний вибір моделі. Хоча кожне нормальне коливання, за визначенням, є коливальним рухом всієї молекули, деякі з них, особливо у великих молекул, можуть найбільше зачіпати лише к.-л. фрагмент молекули. Амплітуди зміщення ядер, що не входять у цей фрагмент, за такого нормального коливання дуже малі. На цьому заснована широко використовується в структурно-аналіт. дослідженнях концепція так званої групових, або характеристичних, частот: певні функціональні групи або фрагменти, що повторюються в молекулах різні сполуки, характеризуються приблизно одними і тими ж частотами в КОЛЕБАТЕЛЬНІ СПЕКТРИ с., за якими може бути встановлена їх присутність у молекулі даної речовини ( правда, який завжди з однаково високим рівнем достовірності). Наприклад, для карбонільної групи характерна дуже інтенсивна смуга в ІЧ спектрі поглинання в області ~1700(b 50) см -1 , що відноситься до валентного коливання . Відсутність смуг поглинання у сфері спектра доводить, що у молекулі досліджуваного речовини групи немає. У той самий час наявність к.-л. смуг у зазначеній області ще не є однозначним доказом присутності в молекулі карбонільної групи, так як у цій галузі можуть випадково виявитись частоти інших коливань молекули. Тому структурний аналіз та визначення конформацій щодо коливальних частот функцій. груп повинні спиратися на кілька характеристик. частот, а передбачувана структура молекули повинна підтверджуватись даними ін. методів (див. Структурна хімія). Існують довідники, які містять численні структурно-спектральні кореляції; є також банки даних та відповідні програми для інформаційно-пошукових систем та структурно-аналіт. досліджень із використанням ЕОМ. Правильної інтерпретації КОЛИВАЛЬНІ СПЕКТРИ с. допомагає ізотопіч. заміщення атомів, що призводить до зміни коливальних частот. Так, заміна водню на дейтерій призводить до зменшення частоти валентного коливання X-Н приблизно 1,4 разу. При ізотопіч. Заміщення силові постійні молекули К е зберігаються. Існує ряд ізотопіч. правил, що дозволяють відносити коливальні частоти до того чи іншого типу симетрії коливань, функціональних груп і т.д. Модельні розрахунки КОЛИВАЛЬНІ СПЕКТРИ с. (частот та інтенсивностей смуг) при заданих силових постійних, які використовують для визначення структури молекул, складають пряме завдання коливальної спектроскопії. Необхідні для цього силові постійні і так звані електрооптичні параметри (дипольні моменти зв'язків, компоненти тензора поляризуемості та ін.) переносять з досліджень близьких за структурою молекул або отримують рішенням зворотного завдання, що полягає у визначенні наборів силових постійних і електрооптичних параметрів багатоатомних молекул. , інтенсивності та ін. Експерим. даних. Визначення наборів фундаментальних частот КОЛИВАЛЬНІ СПЕКТРИ с. необхідно для обчислення коливальних вкладів у термодинамічні функції речовин. Ці дані використовуються в розрахунках хімічної рівноваги та для моделювання технол. процесів. КОЛИВАЛЬНІ СПЕКТРИ с. дозволяють вивчати не тільки внутрімол. динаміку, а й міжмолекулярні взаємодії. З них отримують дані про поверхні потенційної енергії, внутрішньо. обертання молекул, рухи атомів з великими амплітудами. За КОЛИВАЛЬНИМИ СПЕКТРАМИ с. досліджують асоціацію молекул та структуру комплексів різноманітної природи. КОЛИВАЛЬНІ СПЕКТРИ с. залежать від агрегатного стану речовини, що дозволяє отримувати інформацію про структуру різних конденсацій. фаз. Частоти коливальних переходів чітко реєструються на мол. форм з дуже малим часом життя (до 10 -11 с), наприклад, для конформерів при висоті потенційного бар'єру в кілька кДж/моль. Тому КОЛИВАЛЬНІ СПЕКТРИ с. застосовують для дослідження конформаційні ізомерії та швидко встановлюються рівноваг. Про використання КОЛИВАЛЬНІ СПЕКТРИ с. для кількісного аналізу та ін. цілей, а також про сучасну техніку коливальну спектроскопію див. в ст. Інфрачервона спектроскопія, комбінаційне розсіювання спектроскопія.
Поглинання в області 10 2 - 10 3 см -1 (ІЧ - область) обумовлено зазвичай коливальними переходами при постійному електронному стані молекули; відповідні спектри називають коливальними. Точніше їх слід було б назвати коливально-вращательными, т. до. зміна коливальної енергії молекули при поглинанні у цій галузі супроводжується, зазвичай, зміною і обертальної енергії.
h = Е′ – Е″ = (E вр ′ + Е кіль′) – (E вр ″ + Е кіль ″) . (2.104)
Коливальний спектр складається з низки досить далеко віддалених один від одного смуг, інтенсивність яких зі зростанням хвильового числа різко зменшується (рис. 2.22). Першу, найінтенсивнішу смугу називають основною смугою, або основним тоном. Далі розташовуються 1-й та 2-й обертони. Інтенсивність наступних смуг меншає настільки різко, що вже 3-ї та 4-ї обертони для більшості молекул спостерігати не вдається.
Кожна смуга в спектрі є складною і при записі на приладі з великою роздільною здатністю приладу розпадається на ряд окремих ліній. Поява такої тонкої структури притаманна речовин у газоподібному стані. Положення смуг у діапазоні визначається коливальними переходами, а тонка структура кожної лінії – обертальними переходами.
Щоб зрозуміти походження такого спектра, розглянемо спочатку лише коливальний рух і коливальні переходи, абстрагуючись від обертання молекул, тобто.
h = Е кіль - Е кіль . (2.105)
Коливальний рух двоатомної молекули з погляду класичної механіки можна як періодичне зміна відстані між ядрами.

Відповідно до закону Гука, що описує гармонічні коливання,сила, що повертає ядра в положення рівноваги, пропорційна зсуву ядер із положення рівноваги:
f = - kq, (2.106)
де k - Силова постійна;
q – коливальна координата; q = r a + r b = r - r e.
Рівняння Гука справедливе лише для малих зміщень ядер, тобто коли q >> r e , межі при q = 0.
Силова постійна двоатомна молекула є величиною, що характеризує пружність зв'язку і чисельно дорівнює силі, що формує (розтягує або стискає) зв'язок на одиницю довжини f = k при q = 1.
Елементарна робота пружної сили:
dA = - f dq. (2.107)
З урахуванням рівняння (2.106) отримуємо:
dA = - kq dq. (2.108)
Після інтегрування в межах
 (2.109)
(2.109)
для потенційної енергії двоатомної молекули отримуємо:
u = A = 1/2 kq2. (2.110)
З рівняння (2.110) випливає, що
k = (d 2 u / dq 2) q = 0. (2.111)
Таким чином, для малих зсувів потенційна енергія є квадратичною функцією від q = r - r e. Крива u-q або u-r - парабола, а силова постійна k характеризує кривизну параболи поблизу мінімуму.
При підстановці виразу (2.110) рівняння Шредінгера
2 кіль + (8 2 / h 2) (Е кіль – u) кіль = 0 (2.112)
і вирішення цього рівняння виходить наступне рівняння для власних значень коливальної енергії двоатомної молекули як гармонійного осцилятора:
Е кіль = h (v + 1/2) , (2.113)
де v - коливальне квантове числощо приймає значення цілих позитивних чисел, починаючи з нуля (v = 0, 1, 2, 3.......);
0 - Власна частота коливання вібратора.
Рівняння (2.113) можна подати в іншому вигляді:
Е кіль = hc e (v + 1/2) , (2.114)
де e – власне хвильове число (коливальна постійна), що характеризує частоту коливань, віднесену до мінімуму потенційної кривої, тобто ту частоту, яку згідно з класичною механікою мала б молекула для нескінченно малої амплітуди коливань (q = 0, r = re) . Величина e виявляється у м -1 або см -1 . Вона є молекулярною постійною. Будь-яка двоатомна молекула характеризується в кожному електронному стані деяким постійним значенням e.
Рівняння (2.114) вказує на квантування коливальної енергії та існування нульової енергії осцилятора при v = 0:
Е 0 кіл = hc e /2. (2.115)
Ця енергія не дорівнює нулю. Енергія коливань гармонійного осцилятора зростає прямо пропорційно квантовому числу v, що відповідає системі рівновіддалених квантових рівнів. Відповідно до квантово-механічних правил відбору для гармонійного осцилятора можливі переходи з v = 1. При поглинанні світла v змінюється на +1, збільшується енергія та амплітуда коливань.
Однак, модель гармонійного осцилятора призводить до положень, що суперечать експериментальним даним:
1) Екіль у рамках цієї моделі може бути як завгодно великий. У цьому випадку хімічний зв'язок у молекулі був би нескінченно пружним і його розрив був би неможливим. Ми знаємо, що це не так;
2) для гармонійного осцилятора в спектрі поглинання повинна спостерігатися лише одна смуга, що випливає із правил відбору та еквівалентності коливальних рівнів (рис. 2.23 а). Однак у спектрі реальної двоатомної молекули спостерігається кілька смуг.

Рис. 2.23 Криві потенційної енергії (a) та залежність коливальної енергії Е кіль від V кіль (б) для гармонійного осцилятора
Усе це означає, що реальні молекули є гармонічними осциляторами. Гармонійне наближення їм можна використовувати лише за мінімальних зсувах ядер від становища рівноваги, тобто. при малих значеннях коливального квантового числа (v = 0; 1).
Для реальних двоатомних молекул функція U(r) не є параболою, а сила, що повертає, не суворо пропорційна величині зміщення ядер. Це призводить до моделі ангармонічного осцилятора, Для якого крива потенційної енергії зображується, як показано на рис. 2.24.
Для наближеного опису кривої потенційної енергії використовують функцію Морзе:
u = D e 2 (2.116)
де D e - Енергія дисоціації;
– постійна для цієї молекули.

Рис. 2.24 Криві потенційної енергії (а) та залежність коливальної енергії Е кіль від V кіль (б) для ангармонічного осцилятора
При вирішенні рівняння Шредінгера для двоатомної молекули, коли u(r) виражається функцією Морзе, власні значення коливальної енергії Екіл описуються двочленом:
Е кіль = hc e (v +1/2) – hc e x e (v + 1/2) 2 , (2.117)
де х e - Коефіцієнт ангармонічності, що характеризує відхилення від гармонійності, ця величина безрозмірна, причому
e >> e x e > 0. (2.118)
З рівняння (2.117) можна отримати вираз для нульової енергії ангармонічного осцилятора (де v = 0):
Е 0 = 1/2 hc e – 1/4 hc e x e. (2.119)
З рівняння (2.117) випливають висновки:
залежність Екіл від v не є лінійною;
коливальні квантові рівні сходяться зі збільшенням v.
Дійсно, різниця енергії коливань при зростанні квантового числа на одиницю зменшується зі зростанням V:
Е v+1 v = E (v + 1) – E (v) = hc [ e – 2 e x e (v+1)] . (2.120)
Знайдемо першу та другу похідні від функції (2.117):
E v = hc e (V + 1/2) – hc e x e (V + 1/2) 2 , (2.121)
dE V /dV = hc [ e – 2 e x e (V + 1/2)] , (2.122)
d 2 E V /dV = –2hc e x e< 0 . (2.123)
Вираз свідчить у тому, що крива Е v –V має максимум (рис 2.16,б) і коливальні рівні сходяться деякого значення V макс. , Що можна знайти з умови максимуму:
dE V / dV = 0 (2.124)
dE V /dV = hc[ e – 2 e x e (V макс + 1/2)] = 0 , (2.125)
V макс = ( e /2 e x e) – 1/2 , (2.126)
V макс = 1/2x e - 1/2
. (2.127)Таким чином, існує кінцева кількість дискретних коливальних рівнів та максимальна енергія ангармонічного осцилятора Е V, макс. Якщо молекулі повідомити коливальну енергію Е V > EV, макс, відбудеться дисоціація, як це видно з кривої потенційної енергії (рис. 2.16, а).
Розраховані за формулою (2.127) значення V макс для більшості молекул становить кілька десятків, для деяких – до півтори сотні.
Правила відбору:
якщо для гармонійного коливання осцилятора V = 1, то для ангармонічного осцилятора квантово-механічні правила відбору дозволяє будь-які переходи: V = 1, 2, 3 тощо;
можуть бути описані будь-які речовини (полярні та неполярні).
Підставляючи значення V, e , x e рівняння (2.117) можна скласти схему дозволених рівнів енергії коливання.
Рис. 2.25 Схема дозволених рівнів енергій коливань.
Більшість двоатомних молекул коливальний перехід 01 вимагає 10 – 100 кДж/моль. Це значно більше за середню енергію теплового руху молекул газу при температурі 18 – 25 про С (RT = 2,5 кДж/моль при 298 про К). Тому вважатимуться, що з температурі досвіду переважна більшість молекул перебуває в нижньому енергетичному рівні, т. е. V″=0.
Правило відбору дозволяє вивести рівняння для всіх частот, що спостерігаються у спектрі та вивести коливальний спектр:
= EV / hc = e (V + 1/2) – e x e (V + 1/2) 2 . (2.128)
Підставляючи величини V і V в рівняння (2.128) і беручи різницю хвильових чисел отримаємо:
V″ 0 = [ e (V′ + 1/2) – exe (V” + 1/2) 2 ] – [ e (V″ + 1/2) – exe (V″ + 1 /2) 2].(2.129)
Після перетворення:
= (V" – V″) [ e – e x e (1 + V" + V″)] . (2.130)
Враховуючи, що V’=0, отримаємо вираз для хвильових чисел єдино експериментально спостерігається серії переходів, показаних на малюнку, а саме переходів V″ (0)V":
= V" [ e – e x e (1+V")] , (2.131)
де V" = 1, 2, 3, .... V макс.
Найменша енергія потрібна для переходу 01. Це відповідає появі у спектрі поглинання першої (низькочастотної) смуги – основної смуги, або основного типу. Переходи 02; 03 і т. д. дають наступні смуги - обертони.
Хвильові числа основної смуги та обертонів визначається відповідно до (2.131) наступним чином:
01 основна смуга або обертон,
0 1 = e – 2 e x e = e (1 – 2x e), (2.132)
02 1-й обертон,
0 2 = 2 e – 6 e x e = 2 e (1 – 3x e), (2.133)
03 2-й обертон,
0 3 = 3 e – 12 e x e = 3 e (1 – 4x e), (2.134)
У випадку для переходу 0V":
0 V′ = V” e – V′(V′+1) e x e . (2.135)
З отриманих виразів випливає, що смуги поглинання в коливальному спектрі сходяться, хоча через те, що e x e<< e , эта сходимость для первых двух-трех полос выражена слабо. Величина e x e составляет обычно несколько см -1 , реже – десятки см -1 , в то время как e = 10 2 – 10 3 см -1 .
Імовірність переходу 01 найбільша, ніж і пояснюється інтенсивність основної смуги поглинання. Ймовірність переходів 02; 03 і т. д. різко зменшується зі зростанням V", що і відображається в спектрі поглинання.
Визначення коливальної постійної e та коефіцієнта ангармонічностіx e .
Найважливіший результат експериментального вивчення ІЧ спектрів поглинання полягає у визначенні молекулярних постійних – коливальної постійної e та коефіцієнта ангармонічності x e .
відносять смуги поглинання до певних коливальних переходів.
визначають частоту коливань кожного переходу: 1 , 2 , 3 .
зіставляють кожній із частот рівняння типу (2.132) - (2.135) і, вирішивши їх спільно, визначають e і x e . Наприклад:
0 1 = e (1–2x e)
0 2 = 2 e (1–3x e).
Визначення енергії дисоціації (хімічного зв'язку).Енергія хімічного зв'язку є та енергія, яку необхідно витратити, щоб перевести молекулу з нульового на максимальний коливальний квантовий рівень:
Нагадаємо рівняння (2.127):
V макс = 1/2x e - 1/2.
Підставивши це рівняння (2.127), отримаємо:
E 0 Vmax = hc e (1/2x e – 1/2 + 1/2) – hc e x e (1/2x – 1/2 + 1/2) 2 , (2.136)
E 0 Vmax = hc e /2x e – hc e x e /4x e = hc e x e /4x, (2.137)
E max = hc e /4x e. (2.138)
Перейдемо до мольних величин енергії в Дж/моль:
E max (моль) = E max N А, (2.139)
E max (моль) = hc e N А /4x e . (2.140)
Енергію дисоціації, що відраховується від нульового рівня і віднесену до 1молю називають істинною енергією дисоціації та позначають D про:
E х.с. = D o = E max - E 0. (2.141)
Якщо енергію дисоціації відраховувати від мінімуму потенційної кривої, вона перевищує D 0 на величину нульової енергії (рис. 2.18):
D e = D 0 + E 0. (2.142)
hc e NА
hc e

Нагадаємо, що
Е 0 = 1/2 hc e – 1/4 hc e x e ,
D 0 = hc e /4x e – (hc e /2 – hc e x e /4) , (2.143)
D 0 = (1-x e) 2 . (2.144)
Переходячи до мольних величин, знаходимо значення D 0 Дж/моль:
D 0 = (1-x e) 2 . (2.145)
Таким чином: з коливального спектра можна отримати такі молекулярні константи:
Власну частоту коливань е;
Коефіцієнт ангармонічності хе;
Енергію коливального руху молекул;
Енергію хімічного зв'язку.
Електронні діапазони (основні поняття).При збудженні електронів у молекулах спостерігається випромінювання в ультрафіолетовій та видимій областях спектра.
h = E"" – E" = (E"" вр – E" вр) + (E" "кіль – E" кіл) + (E" "ел – E" ел).
П  При цьому має місце сукупність усіх видів енергетичних змін. Спектр складний і називається електронно-коливально-обертальним. Спектр складається із смуг поглинання. Максимум лінії поглинання відповідає найбільш можливому переходу в даній області довжин хвиль.
При цьому має місце сукупність усіх видів енергетичних змін. Спектр складний і називається електронно-коливально-обертальним. Спектр складається із смуг поглинання. Максимум лінії поглинання відповідає найбільш можливому переходу в даній області довжин хвиль.
На малюнку 2.25 показано відносне розташування енергетичних рівнів молекулярних орбіталей МО ( і – зв'язуючі МО, * та * – розпушують МО)
В основному стани - та -орбіталі зазвичай зайняті електронами (це більш стійкий енергетичний стан з меншою потенційною енергією).
Найбільшої енергії вимагає перехід * – виявляється у дальній УФ-області і уражає молекул насичених вуглеводнів. Переходи * відповідають видимій та ближній УФ-областям та типові для молекул ненасичених сполук.

Рис. 2.26. Криві потенційної енергії взаємодії при електронних переходах 
При поглинанні великих квантів променистої енергії може статися електронний перескок. Потенційна енергія дисоціації D0 – зменшується, а Е – збільшується. У разі підвищення енергії Е збільшується міжатомна відстань r e в результаті коливального руху (рис 2.26).
Для кожного виду зв'язку є своя енергія електронних переходів і характерна смуга поглинання з певною довжиною хвилі.